
The medtech industry is undergoing significant change, from the introduction of the new MDR and the update to ISO 13485, to the growth of home-based healthcare and the uncertainties of Brexit.
This post was originally published in November 2017 and updated in March 2018.
At some recent Irish medtech events, the major topics dominating conversation were:
- Regulation 2017/745 on Medical Devices – widely known as the MDR
- Usability and the importance of the end user
Indeed, these topics are interrelated, given that the central aim of the MDR is to establish high standards of usability and avoid safety risks and hazards for end users.
Unfortunately, insufficient attention is being given to documentation in the current discussion, despite its central importance to the MDR and the experience of the end user. For end users, labelling and Instructions for Use (IFUs) are key to learning about using a medical device, and understanding its functions and limits. The quality of the documentation that users receive can often dictate whether their experience of using the device is a positive or negative one. Manufacturers are also required to implement post-market surveillance of their products and conduct a Summary of Safety and Clinical Performance (SSCP). These processes are also required to be documented.
This post primarily focuses on documentation, examining the role it plays in optimising usability, enhancing user experience, and supporting MDR compliance.

Are You Preparing for the MDR?
By now, we’re probably all familiar with the term “MDR”. The MDR is new legislation from the European Commission that officially came into force on 26 May 2017.*
The medtech industry is currently in the implementation phase, or, as it is commonly described, a transition from the former Medical Device Directives (MDDs) to the MDR. Medtech companies are tasked with updating their documentation and processes by May 2020 – the point at which all companies must be fully compliant with the MDR.
Key aspects of the MDR include changes in device classifications, new requirements around clinical data and post-market surveillance, and the introduction of Unique Device Identifiers (UDIs). Many medtech organisations are currently in the process of investigating how the MDR affects their business, so that they can prepare an implementation plan. This is a challenge for enterprises of all sizes, but particularly for small medtech companies and start-ups with limited resources.
In addition, the Quality Management System ISO 13485 was updated in 2016. Manufacturers of medical devices have until March 2019 to update their certificate. The MDR and ISO 13485 iterate similar guidelines, most notably, the requirement for post-market surveillance.
User Documentation and the MDR
The introduction of the MDR brings more clarity in terms of medical device regulations and improvements in patient treatment and user safety and experience. The MDR focuses heavily on safeguarding the end user and ensuring that they are suitably well-informed about devices. Therefore, it is unsurprising that the regulation affords documentation (and, particularly, user documentation) a key role.
The MDR sets out the requirements regarding the information supplied by manufacturers to medical device users. Each device must be accompanied by information specifying all performance and safety information relevant to the user. Clear usage and disposal procedures must be provided for devices, and content must be tailored to the level of the target audience’s technical knowledge.
The regulation states:
The medium, format, content, legibility, and location of the label and instructions for use shall be appropriate to the particular device, its intended purpose and the technical knowledge, experience, education or training of the intended user(s). In particular, instructions for use shall be written in terms readily understood by the intended user and, where appropriate, supplemented with drawings and diagrams.
In addition, ISO 13485 outlines the requirement for a medical device file. The file should include the following documents:
- General description
- Intended use or purpose
- Labelling and IFU
- Specifications for the product
- Instructions for manufacturing, packaging, storage and distribution
- Procedures for measuring and monitoring
- Installation requirements.as required
- Servicing requirements, as required
ISO 13485 and the MDR both outline other processes intended to improve the quality and thus usability of medical devices, that must also be documented. Article 83 of Chapter VII describes the requirement for a post-market surveillance system of the manufacturer. The system is used to analyse the performance of a medical device and thus draw up any preventative and corrective actions required. Section 8.1 of ISO 13485 addresses the necessity of post-market surveillance. The plan and any reports developed during implementation of the plan are to be documented.
Similarly, Article 32 describes how manufacturers of implantable devices and class III devices, other than custom-made or investigational devices, shall draw up a summary of safety and clinical performance. Again, the summary should be understandable to the end user and should be made available on the EUDAMED online database.
This central focus on the end user parallels the growing emphasis on usability that is evident throughout the medtech field.
Importance of the End User
The medtech industry is moving towards consensus on the centrality of the end user to product design and development. Medical devices need to be easy to use to succeed in a market where users wield increasing power. Whether end users are patients, home caregivers, or medical professionals, they expect medtech companies to offer more personalised products, easier procedures, and an enhanced user experience.
Satisfying these end users requires a multidisciplinary approach involving engineering, design, and surveillance, among others. Currently, there is a move from a product to a service model for medical devices and greater end user involvement in every stage of product development. In future, therefore, medtech development will need to rely on a much broader range of inputs, together with extensive real-world data and usability feedback.
At the Medical Technology Ireland Expo and Conference 2017 and the MedTec Ireland 2017 Conference, speakers and delegates agreed that investment in user-centred development is vital for successful product development. However, they cautioned that this must not come at the cost of investment in supply chain and manufacturing, and process improvement initiatives. All of these elements form part of the usability chain, and have a role in producing a safe and effective product for end users. Greater usability will not be achieved at the cost of process improvement efforts, but rather through their implementation. Process improvement is another area where effective documentation can aid successful outcomes.
User-Centred Documentation Strategy
Documentation plays a crucial role in optimising users’ experience of a product. The prioritisation of the end user evident throughout the medtech industry underlines the importance of telling the story of products effectively for users in marketing material, and providing clear, user-centred documentation. User documentation is a vital part of the usability chain – a chain that extends across the product life cycle from initial Research and Development (R&D) through design and post-market surveillance activities.
Given the MDR implementation time frame, the need to create accessible, end-to-end documentation of the following is an immediate reality for medtech companies.
- IFU and labelling
- The post-market surveillance plan and any findings of that plan
- Medical device file
- SSCP
It is essential for companies to consider whether they have the available in-house skills and resources to achieve the required standards of user documentation. If not, their options are to:
- Provide training to help enhance the writing skillset of employees tasked with documentation activities outside of their main role
- Avail of the support of documentation experts in developing clear instructions and user-friendly content
End-To-End Documentation
To succeed in the new regulatory climate, and the increasingly user-led marketplace, medtech companies need to acknowledge the important role of documentation in the usability chain, and consider how the various types of documentation involved in product development can support business processes and optimise usability.
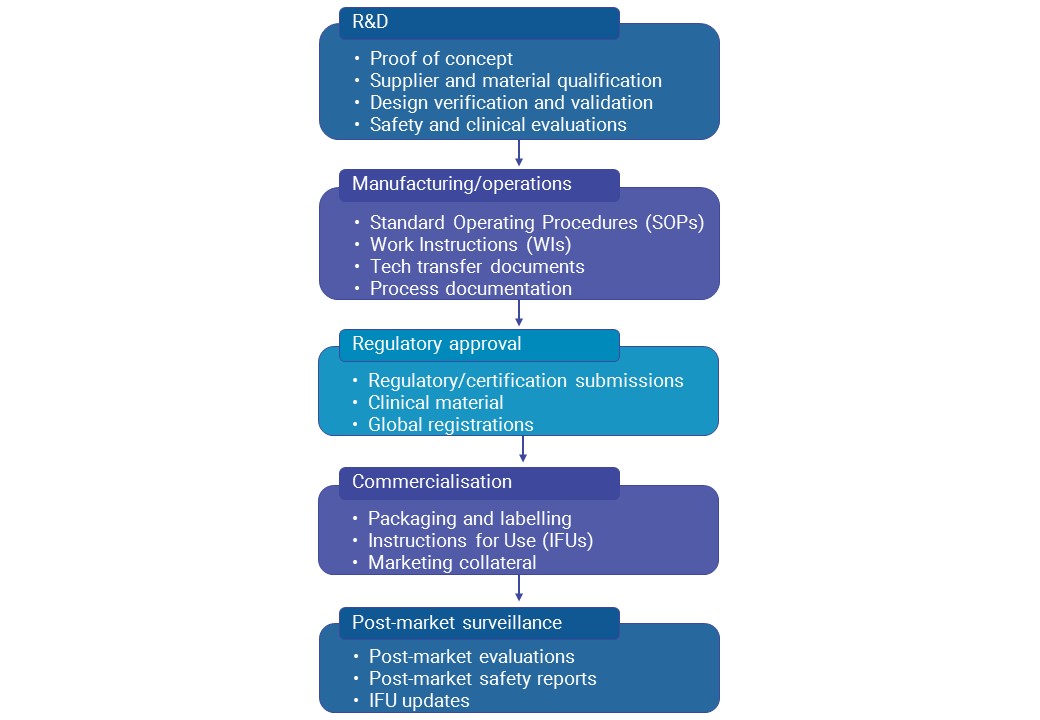
Overview of documentation requirements throughout the medtech product lifecycle.
Implementing a coherent, end-to-end documentation strategy will help companies to:
- Meet their regulatory obligations
- Support end user safety and satisfaction
- Facilitate greater organisational efficiency and foster process improvement
In short, careful end-to-end documentation planning can support process refinement across the entire product life cycle, while promoting better user outcomes.
The essential first step is to recognise the central role of documentation in achieving MDR compliance, as well as process and usability excellence. But where do you go from there? We recommend carrying out an end-to-end audit of your organisation’s current documentation practices. You’ll gain a clear picture from this analysis, allowing you to identify gaps, weaknesses, and areas for improvement.
Images Used
- Image by Hush Naidoo, licensed by Unsplash
- Image by Hush Naidoo, licensed by Unsplash
HAVE SOMETHING TO SAY?
If you’d like to share your thoughts or know more about this topic, complete our feedback form. We look forward to hearing from you!










